|
Cloudy
Spectral Synthesis Code for Astrophysics
|
|
Cloudy
Spectral Synthesis Code for Astrophysics
|
#include <mole.h>
Public Types | |
| typedef vector< shared_ptr < molecule > > | MoleculeList |
Public Member Functions | |
| const char * | chName () const |
| void | zero () |
| void | comment (t_warnings &) |
| void | init (void) |
| void | make_species (void) |
 Public Member Functions inherited from module Public Member Functions inherited from module | |
| module () | |
| virtual | ~module () |
Static Public Member Functions | |
| static void | sort (MoleculeList::iterator start, MoleculeList::iterator end) |
| static void | sort (molecule **start, molecule **end) |
Public Attributes | |
| bool | lgNoMole |
| bool | lgNoHeavyMole |
| bool | lgH2Ozer |
| bool | lgLeidenHack |
| bool | lgFederman |
| bool | lgStancil |
| bool | lgNonEquilChem |
| bool | lgProtElim |
| bool | lgNeutrals |
| bool | lgGrain_mole_deplete |
| vector< bool > | lgTreatIsotopes |
| int | num_total |
| int | num_calc |
| int | num_compacted |
| MoleculeList | list |
| map< string, bool > | offReactions |
| map< string, Properties > | speciesProperties |
| typedef vector<shared_ptr<molecule> > t_mole_global::MoleculeList |
|
inlinevirtual |
Implements module.
|
inlinevirtual |
Implements module.
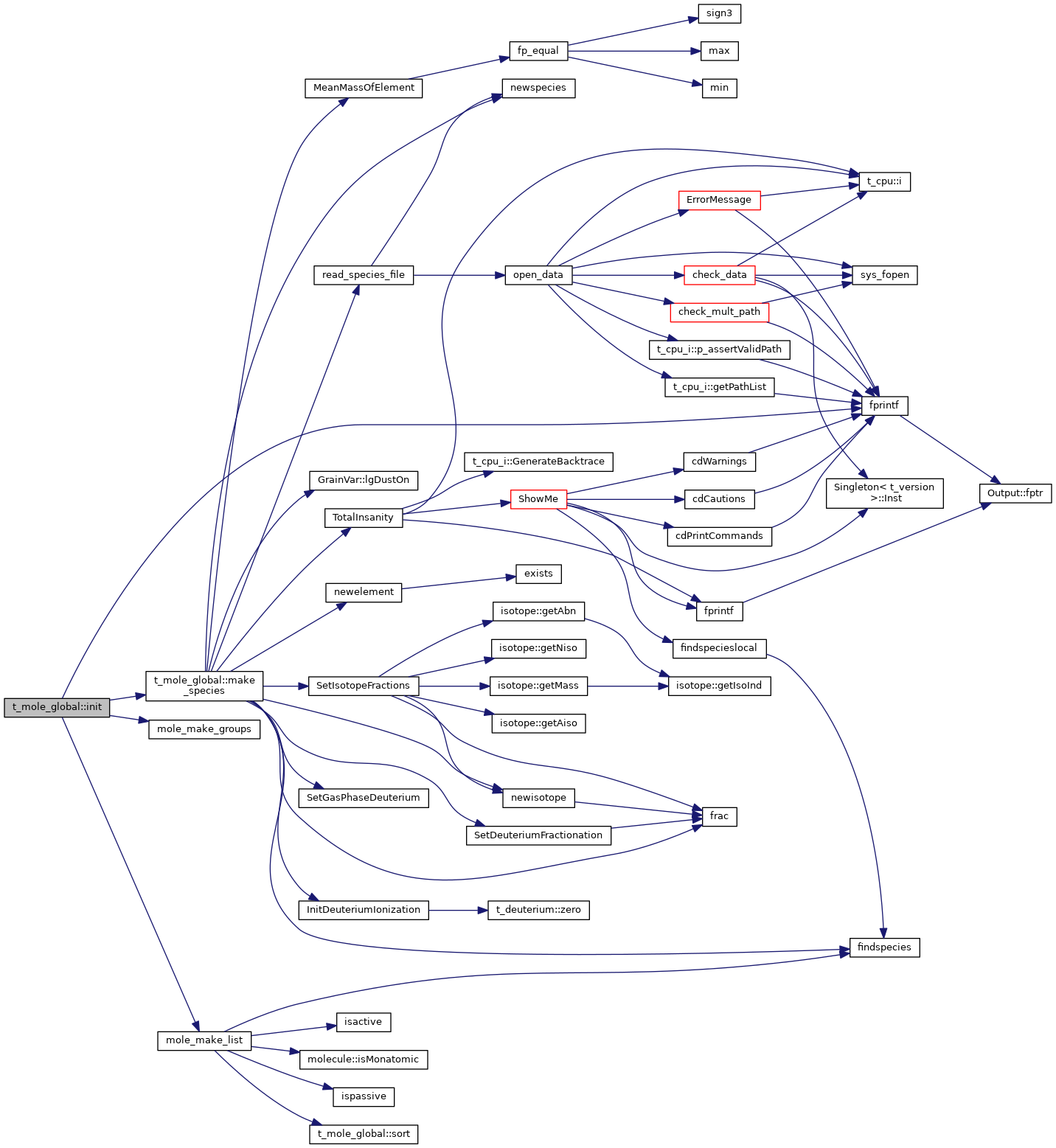
| void t_mole_global::init | ( | void | ) |
mole_zero allocate + initialize workspace
References cdEXIT, DEBUG_ENTRY, t_mole_local::elec, EXIT_FAILURE, fprintf(), ioQQQ, make_species(), mole_make_groups(), mole_make_list(), num_total, and t_mole_local::species.
Referenced by InitSimPostparse().
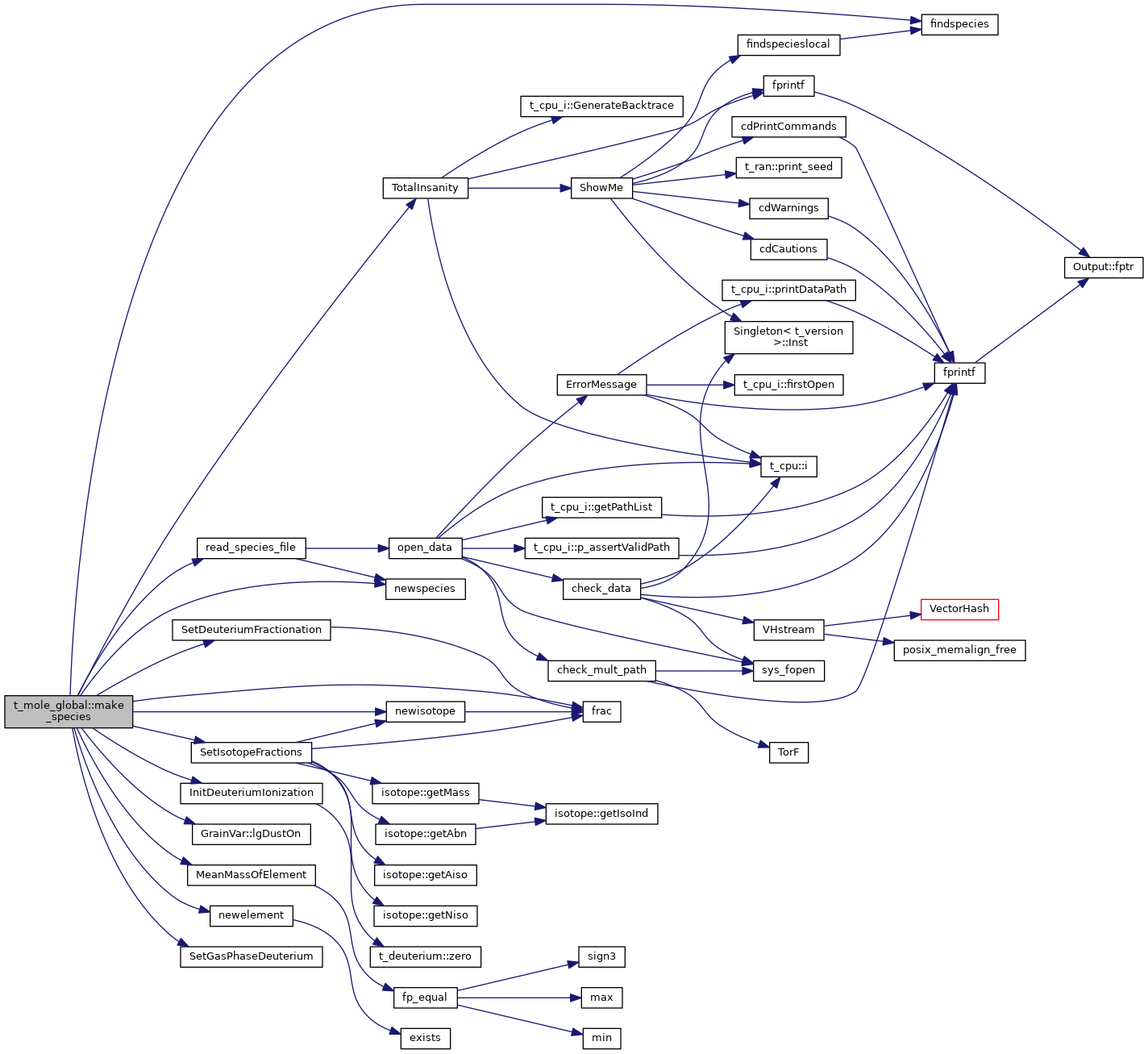
| void t_mole_global::make_species | ( | void | ) |
References molecule::charge, CHARS_ION_STAGE, CHARS_ISOTOPE_SYM, t_elementnames::chElementSym, DEBUG_ENTRY, molezone::den, dense, deut, element_list, elementnames, diatomics::ENERGY_H2_STAR, findspecies(), fixit, frac(), t_dense::gas_phase, gv, h2, hmi, molecule::index, InitDeuteriumIonization(), ipHYDROGEN, GrainVar::lgDustOn(), t_deuterium::lgElmtOn, lgGrain_mole_deplete, t_hmi::lgLeiden_Keep_ipMH2s, lgLeidenHack, lgTreatIsotopes, LIMELM, MeanMassOfElement(), MOLE_ACTIVE, mole_global, molecule::mole_mass, MOLE_NULL, MOLE_PASSIVE, MOLECULE, newelement(), newisotope(), newspecies(), nuclide_list, NULL, null_mole, null_molezone_impl, null_nuclide_impl, num_calc, num_total, OTHER, read_species_file(), SetDeuteriumFractionation(), SetGasPhaseDeuterium(), SetIsotopeFractions(), and TotalInsanity().
Referenced by init().
|
static |
Referenced by canonicalize_reaction(), mole_make_list(), and parse_udfa().
|
virtual |
option to set proton elimination rates to zero >>refer CO chemistry Huntress, W. T., 1977, ApJS, 33, 495 By default, this is false - changed with set chemistry command
option to not include neutrals in the non-equilibrium scheme
refer Federman, S. R. & Zsargo, J. 2003, ApJ, 589, 319
By default, this is false - changed with set chemistry command
Implements module.
References DEBUG_ENTRY, lgFederman, lgGrain_mole_deplete, lgH2Ozer, lgLeidenHack, lgNeutrals, lgNoHeavyMole, lgNoMole, lgNonEquilChem, lgProtElim, lgStancil, lgTreatIsotopes, and LIMELM.
| bool t_mole_global::lgFederman |
option to use diffuse cloud chemical rates from Table 8 of
refer Federman, S. R. & Zsargo, J. 2003, ApJ, 589, 319
By default, this is true - changed with set chemistry command these rates are in both mole_co_federman.dat and mole_co_base.dat, latter based on UMIST
Referenced by mole_create_react(), ParseSet(), and zero().
| bool t_mole_global::lgGrain_mole_deplete |
do we include capture of molecules onto grain surfaces? default is true, turned off with NO GRAIN MOLECULES command
Referenced by make_species(), ParseDont(), and zero().
| bool t_mole_global::lgH2Ozer |
flag set true if H2O destruction rate went to zero
Referenced by PrtComment(), and zero().
| bool t_mole_global::lgLeidenHack |
option to turn on the original UMIST rates rather than the corrected network adopted in the Leiden PDR meeting, by deffault false, set true with the set UMIST rates command, these rates are from mole_co_umisthack.dat and also in mole_co_base.dat (UMIST)
Referenced by ChargTranEval(), ion_photo(), make_species(), mole_create_react(), mole_h_reactions(), ParseSet(), radius_next(), and zero().
| bool t_mole_global::lgNeutrals |
option to not include neutrals in the non-equilibrium scheme
refer Federman, S. R. & Zsargo, J. 2003, ApJ, 589, 319
By default, this is false - changed with set chemistry command
Referenced by ParseSet(), and zero().
| bool t_mole_global::lgNoHeavyMole |
flag to turn off heavy molecules, set with no heavy molecules command
Referenced by newspecies(), ParseDont(), and zero().
| bool t_mole_global::lgNoMole |
flag to turn off all molecules, set with no molecules command
Referenced by ConvInitSolution(), CoolEvaluate(), ion_trim(), ion_trim2(), newspecies(), ParseCommands(), ParseDont(), and zero().
| bool t_mole_global::lgNonEquilChem |
option to use effective temperature as defined in
refer Federman, S. R. & Zsargo, J. 2003, ApJ, 589, 319
By default, this is false - changed with set chemistry command
Referenced by ParseSet(), and zero().
| bool t_mole_global::lgProtElim |
option to set proton elimination rates to zero >>refer CO chemistry Huntress, W. T., 1977, ApJS, 33, 495 By default, this is false - changed with set chemistry command
Referenced by mole_create_react(), ParseSet(), and zero().
| bool t_mole_global::lgStancil |
| vector<bool> t_mole_global::lgTreatIsotopes |
Referenced by make_species(), newspecies(), ParseElement(), and zero().
| MoleculeList t_mole_global::list |
Referenced by t_gaunt::brems_sum_ions(), ChemImportance(), ConvBase(), ConvIterCheck(), DynaStartZone(), eden_sum(), ElectronFractions(), funjac(), getMolecules(), molezone::global(), isSpeciesActive(), iter_end_check(), lgNucleiConserved(), matchGeneric(), molcol(), mole_cmp_num_in_out_reactions(), mole_eval_balance(), mole_eval_dynamic_balance(), mole_h_rate_diagnostics(), mole_make_groups(), mole_make_list(), mole_return_cached_species(), mole_update_limiting_reactants(), mole_update_species_cache(), PrtComment(), PrtZone(), radius_next(), SaveDo(), SaveSpecies(), test_isMolecule(), total_molecule_deut(), total_molecule_elems(), total_molecules(), total_molecules_gasphase(), and GroupMap::updateMolecules().
| int t_mole_global::num_calc |
Referenced by t_gaunt::brems_sum_ions(), ChemImportance(), ConvBase(), ConvIterCheck(), DynaCreateArrays(), DynaIonize(), DynaNewStep(), DynaSaveLast(), DynaStartZone(), eden_sum(), ElectronFractions(), funjac(), init_struc(), iter_end_check(), IterRestart(), IterStart(), lgNucleiConserved(), make_species(), molcol(), mole_eval_balance(), mole_eval_dynamic_balance(), mole_h_fixup(), mole_h_rate_diagnostics(), mole_make_groups(), mole_make_list(), mole_return_cached_species(), mole_solve(), mole_update_limiting_reactants(), newspecies(), PrtComment(), PrtZone(), radius_increment(), radius_next(), RT_tau_reset(), SaveDo(), total_molecule_deut(), total_molecule_elems(), total_molecules(), total_molecules_gasphase(), GroupMap::updateMolecules(), and t_mole_local::zero().
| int t_mole_global::num_compacted |
Referenced by funjac(), grouped_elems(), mole_make_groups(), mole_solve(), GroupMap::setup(), and GroupMap::updateMolecules().
| int t_mole_global::num_total |
flag saying whether an element is in the chemistry network
Referenced by AbundChange(), atmdat_readin(), funjac(), init(), make_species(), mole_cmp_num_in_out_reactions(), mole_effects(), mole_make_list(), mole_return_cached_species(), mole_update_sources(), mole_update_species_cache(), newspecies(), plot_sparsity(), ScaleAllDensities(), t_mole_local::set_ion_locations(), and GroupMap::setup().
| map<string,bool> t_mole_global::offReactions |
Referenced by mole_create_react(), newreact(), and ParseChemistry().
| map<string,Properties > t_mole_global::speciesProperties |
Referenced by ParseSpecies(), setProperties(), speciesCheck(), and speciesOff().
 1.8.5
1.8.5 
